DISTROFIA MUSCULAR DE DUCHENNE
El cambio en el gen que provoca la distrofia muscular de Duchenne o de Becker (DMDB) se produce en el cromosoma X. Un niño hereda un cromosoma X de su madre y un cromosoma Y de su padre. Solo el cromosoma X puede tener el gen modificado que provoca la DMDB. Las mujeres casi nunca presentan DMDB, porque tienen dos cromosomas X. Incluso, si una mujer tiene un cromosoma X con el gen de la DMDB, su segundo cromosoma X, generalmente, producirá suficiente distrofina para mantener los músculos fuertes.
¿Qué son las distrofias musculares?
Las distrofias musculares son muchos trastornos hereditarios del músculo esquelético que tienen en común un daño muscular progresivo que normalmente se manifiesta entre la infancia y la edad adulta.
Las distrofias musculares más frecuentes están ligadas al cromosoma X y están causadas por mutaciones que alteran la función de una proteína estructural grande denominada distrofina. Por esta razón, estas enfermedades a veces se denominan distrofinopatías. La forma de inicio temprano más frecuente es la distrofia muscular de Duchenne. Tiene una incidencia de 1 caso por 3.500 nacidos vivos de sexo masculino y un fenotipo progresivo grave. Igual que muchas enfermedades ligadas al cromosoma X, las mujeres portadoras de mutaciones de la distrofina pueden presentar síntomas leves por una desactivación desfavorable del cromosoma X.
¿Cuál es su patogenia?
La distrofia muscular de Duchenne está causada por mutaciones con pérdida de función en el gen de la distrofina del cromosoma X. El gen de la distrofina es uno de los genes humanos más grandes, con 2,3 millones de pares de bases y 79 exones.
La distrofia muscular de Duchenne se asocia, por lo general, a deleciones o mutaciones con desplazamiento del marco de lectura que causan una ausencia total de distrofina.
Hablemos de su morfología
Esta enfermedad se caracteriza por un daño muscular crónico que sobrepasa la capacidad de reparación ( fig. 1 ). La biopsia muscular en niños pequeños muestra daño progresivo en forma de degeneración y regeneración segmentarias de la miofibra asociado a una mezcla de miofibrillas atróficas. La estructura fascicular se conserva en esta fase de la enfermedad y, por lo general, no hay inflamación, excepto por la presencia de miofagocitosis. Al avanzar la enfermedad, el tejido muscular se sustituye por colágeno y células grasas («sustitución grasa» o «infiltración grasa»). En esta fase de la evolución, las miofibrillas restantes presentan una notable variedad de tamaño, desde fibras atróficas pequeñas hasta fibras hipertróficas grandes. Es decir, la remodelación distorsiona la estructura fascicular del músculo, que se altera considerablemente con el tiempo.
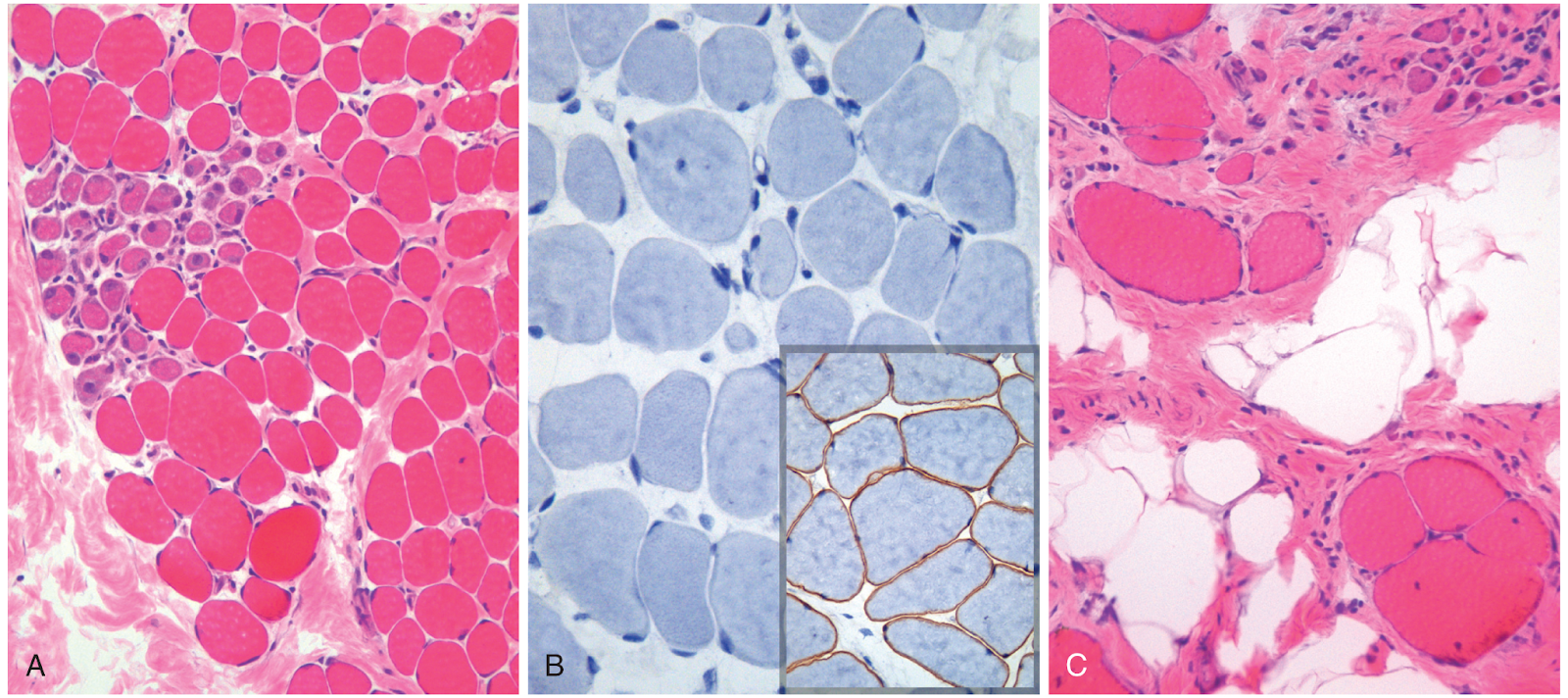
Figura 1: Distrofia muscular de Duchenne. Imágenes histológicas de la biopsia muscular de dos hermanos. A y B. Imágenes de un niño de 3 años. C. Imagen de su hermano de 9 años. A. A una edad más temprana se mantiene la estructura muscular fascicular, pero se observa variedad de tamaño de las miofibrillas. Además, hay un conglomerado de miofibrillas en regeneración basófilas (lado izquierdo) y ligera fibrosis endomisial, que se observa como tejido conjuntivo focal con tinción rosa entre las miofibrillas. B. La tinción inmunohistoquímica muestra ausencia total de distrofina asociada a la membrana, que se manifiesta por una tinción marrón en el músculo normal (recuadro). C. La biopsia del hermano mayor muestra el avance de la enfermedad, que se caracteriza por una variación extensa del tamaño de las miofibrillas, sustitución grasa y fibrosis endomisial.
Características clínicas
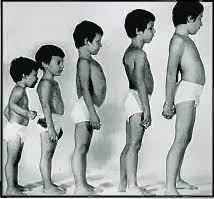
Los niños con distrofia muscular de Duchenne parecen normales al nacer. Consiguen los primeros logros de habilidad motora, pero, a menudo, tardan en empezar a andar. Los primeros indicios de debilidad muscular son la torpeza y la incapacidad para seguir el ritmo de los niños sanos. La debilidad comienza en los músculos de la cintura pélvica y después se extiende a la cintura escapular. Muchos de estos niños presentan un aumento de tamaño de los músculos de la pantorrilla asociado a la debilidad, denominado seudohipertrofia. La media de edad de dependencia de la silla de ruedas está alrededor de 9,5 años. Los pacientes presentan contracturas musculares, escoliosis, empeoramiento de la reserva respiratoria e hipoventilación al dormir.
La distrofina se expresa también en el corazón y en el SNC. La deficiencia de distrofina en el músculo cardíaco ocasiona con frecuencia miocardiopatía y arritmias, sobre todo en los pacientes mayores. También es frecuente el deterioro cognitivo y las dificultades de aprendizaje, supuestamente por el papel funcional de la distrofina en el encéfalo, y a veces produce una discapacidad intelectual patente. A pesar del tratamiento complementario, la media de edad a la que mueren los pacientes con distrofia muscular de Duchenne es de 25-30 años, la mayoría por insuficiencia respiratoria, infección pulmonar o insuficiencia cardíaca.

Diagnóstico
Se basa en la anamnesis, la exploración física y los análisis clínicos. La creatina cinasa está muy elevada durante la primera década de la vida por el daño muscular persistente, y después desciende al avanzar la enfermedad y perder masa muscular. La detección de una mutación de la distrofina proporciona un diagnóstico definitivo.


Tratamiento
El tratamiento actual consiste, principalmente, en medidas complementarias. El tratamiento definitivo consiste en normalizar la concentración de distrofina en las fibras musculares esqueléticas y cardíacas. Los trabajos en este campo se han visto incentivados por el reconocimiento de que la expresión parcial de la proteína distrofina (como en los pacientes con distrofia muscular de Becker) es suficiente para mejorar bastante el fenotipo de la enfermedad. Una estrategia consiste en la expresión de varios ARN antisentido, que alteran el proceso de empalme de ARN para conseguir «saltarse» los exones que contienen mutaciones perjudiciales, permitiendo así la expresión de una proteína distrofina truncada, pero parcialmente funcional. Otro método es utilizar fármacos que estimulan la «lectura previa» ribosómica de los codones de detención, otra estrategia que puede conseguir la expresión parcial de proteína distrofina. Ambos métodos son específicos de la mutación y, por tanto, deben adaptarse a los pacientes de manera individualizada. Está investigándose la terapia génica (introducción de un gen de distrofina normal), pero sigue siendo muy difícil hacer llegar un gen a las células del músculo esquelético.
A continuación se muestra un testimonio de un paciente narrando cómo es vida y experiencia con ésta enfermedad:
https://www.youtube.com/watch?v=hzA4ur09eOs
¿Cómo se hereda la distrofia muscular de Duchenne?
Debido a que una mujer puede portar (o tener) una mutación de la DMDB y no verse afectada, se la denomina portadora. Como portadora, la mujer corre el riesgo de transmitir la misma mutación a sus hijos. Cada hijo de una mujer portadora tiene una posibilidad del 50 % de heredar la mutación de la DMDB y de presentar DM. Cada hija de una mujer portadora, por otro lado, tiene una posibilidad del 50 % de heredar la mutación de la DMDB y de convertirse en portadora como su madre.
Si bien se conoce que muchos hombres diagnosticados con DMDB han heredado la mutación de sus madres, alrededor de un tercio de los casos son el resultado de una nueva mutación en la que la madre no es portadora. En cambio, la nueva mutación apareció de forma aleatoria en el óvulo fertilizado. En estos casos, es poco probable que otros niños de esa misma pareja se vean afectados de forma similar por la DMDB.
Para comprender mejor este proceso de herencia se recomienda ver el siguiente video:
https://www.youtube.com/watch?v=4vYZY7MEGks
A continuación se enlaza un artículo que explica a mayor detalle esta enfermedad.
Referencias:
Kumar, V., Abbas, A. y Aster, J. (2021). ROBBINS Y COTRAN. PATOLOGÍA ESTRUCTURAL Y FUNCIONAL. DÉCIMA EDICIÓN. ELSEVIER.
CDC (s.f.). Cómo se hereda la distrofia muscular de Duchenne o de Becker. Recuperado el 16 de febrero de 2024, de https://www.cdc.gov/ncbddd/spanish/musculardystrophy/inheritance.html
Duchenne y Tú (2020). Entender la genética de la distrofia muscular de Duchenne. [Video]. https://www.youtube.com/watch?v=4vYZY7MEGks
Azteca Noticias (2023). Enfermedades raras y sin cura: ¿Qué es la Distrofia muscular de Duchenne?. [Video]. https://www.youtube.com/watch?v=hzA4ur09eOs
Camacho, A. (2014). Distrofia muscular de Duchenne. Anales de Pediatría Continuada, 12(2), 47-54. https://www.elsevier.es/es-revista-anales-pediatria-continuada-51-articulo-distrofia-muscular-duchenne-S1696281814701684
Teletón (s.f.). DISTROFIA MUSCULAR DE DUCHENNE. Recuperado el 16 de febrero de 2024, de https://teleton.org/distrofia-muscular-de-duchenne/
Elaboró: Martinez Yañez Melisa


Muy interesnate tema, me gustaría complementar que otras formas de diagnóstico son una biopsia muscular o un estudio genético, en donde en la biopsia se observa un patrón distrófico característico y la inmunohistoquímica proporciona el diagnóstico definitivo al demostrar ausencia de tinción de la distrofina.
ResponderBorrar-Juan Manuel Zamora Hernández